PubMed
Full publications list.
Selected Publications
2025 Publications
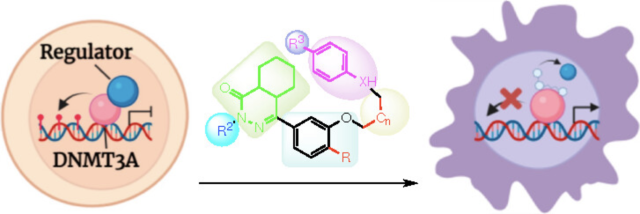
Allosteric Inhibitors of Cell-Cycle-Regulated Methyltransferase for Novel Antibiotic Development
Ivan Hernandez, Kyongyun Claire Jin, Yicheng Yang, Olivia Konttinen, Alexandra Lantz, Yifan Zhao, Ian Squire, Thomas R. R. Pettus, and Norbert O. Reich. ACS Omega 2025 10 (15), 15775-15780
DOI: 10.1021/acsomega.5c01540
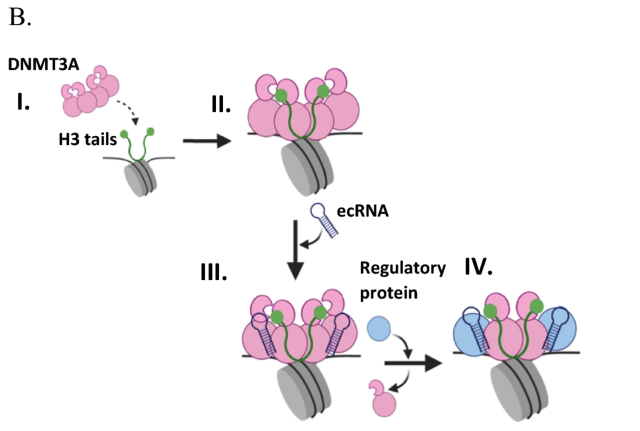
Mechanism of non-coding RNA regulation of DNMT3A
J. E. Sandoval, N. V. N. Carullo, A. J. Salisbury, J. J. Day and N. O. Reich. Epigenetics & Chromatin 2025 Vol. 18 Issue 1 Pages 15
https://doi.org/10.1186/s13072-025-00574-w
2024 Publications
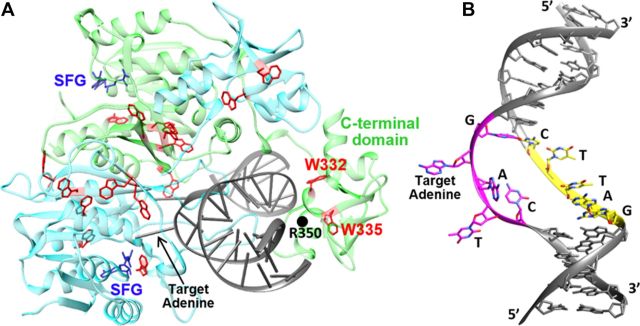
Structural Investigations of Phthalazinone Derivatives as Allosteric Inhibitors of Human DNA Methyltransferase 3A
Ivan Hernandez, Ethan Ward, Thomas R. R. Pettus, and Norbert O. Reich. ACS Medicinal Chemistry Letters 2024 15 (5), 590-594
DOI: 10.1021/acsmedchemlett.3c00528
2023 Publications
High-fidelity DNA strand separation is the major specificity determinant in DNA methyltransferase CcrM's catalytic mechanism.
Konttinen, Olivia & Carmody, Jason & Kurnik, Martin & Johnson, Kenneth & Reich, Norbert. Nucleic acids research, 51(13) (2023).
DOI: https://doi.org/10.1093/nar/gkad443

2022 Publications
In silico Study of Selective Inhibition Mechanism of S-Adenosyl-L-Methionine Analogs for Human DNA Methyltransferase 3 A.
Stillson, Nathaniel & Anderson, Kyle & Reich, Norbert. Computational Biology and Chemistry. 102(17) (2022).
10.1016/j.compbiolchem.2022.107796
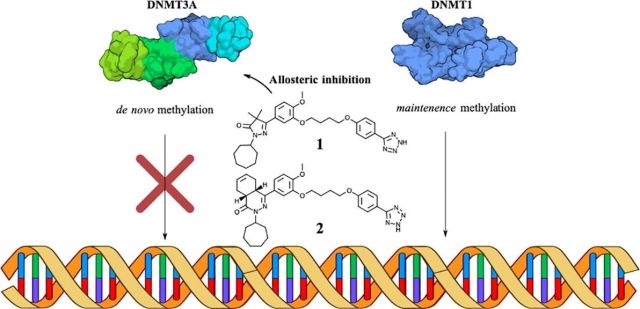
First-in-Class Allosteric Inhibitors of DNMT3A Disrupt Protein–Protein Interactions and Induce Acute Myeloid Leukemia Cell Differentiation.
Sandoval, Jonathan & Ramabadran, Raghav & Stillson, Nathaniel & Sarah, Letitia & Fujimori, Danica & Goodell, Margaret & Reich, Norbert. Journal of Medicinal Chemistry. 65(15) (2022).
10.1021/acs.jmedchem.2c00725
2021 Publications
p53 and TDG are dominant in regulating the activity of the human de novo DNA methyltransferase DNMT3A on nucleosomes
Sandoval JE, Reich NO. J. Biol. Chem., 296 (2021), 100058
https://doi.org/10.1074/jbc.RA120.016125
A novel class of selective non-nucleoside inhibitors of human DNA methyltransferase 3A
Huang S, Stillson NJ, Sandoval JE, Yung C, Reich NO. Bioorg. Med. Chem. Lett., 40 (2021), 127908
https://doi.org/10.1016/j.bmcl.2021.127908
2020 Publications
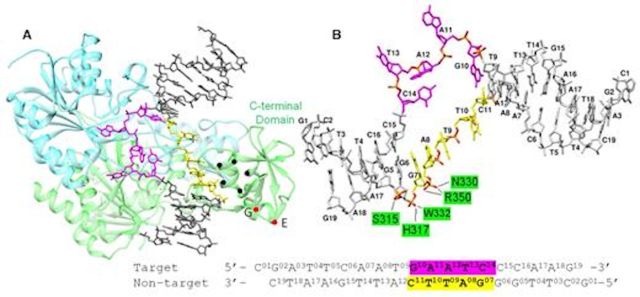
Cell cycle regulated DNA methyltransferase: fluorescent tracking of a DNA strand-separation mechanism and identification of the responsible protein motif
Konttinen O, Carmody J, Pathuri S, Anderson K, Zhou X, Reich NO. Nucleic Acids Res., 48(20) (2020), 11589-11601
https://doi.org/10.1093/nar/gkaa844
2019 Publications
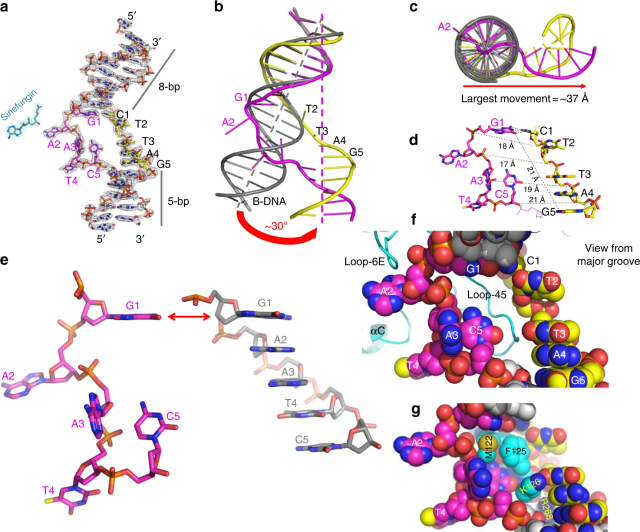
The cell cycle-regulated DNA adenine methyltransferase CcrM opens a bubble at its DNA recognition site
Horton JR, Woodcock CB, Opot SB, Reich NO, Zhang X, Cheng X. Nat. Commun., 10(1) (2019), 4600
https://doi.org/10.1038/s41467-019-12498-7
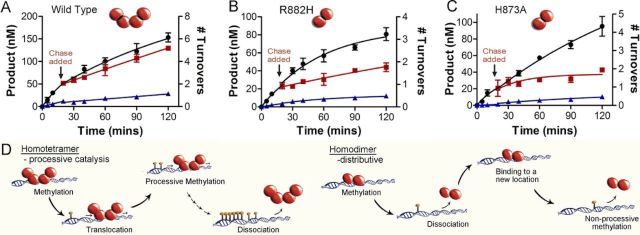
The R882H substitution in the human de novo DNA methyltransferase DNMT3A disrupts allosteric regulation by the tumor supressor p53
Sandoval JE, Reich NO. J. Bio. Chem., 294(48) (2019), 18207-18219
https://doi.org/10.1074/jbc.RA119.010827
Mutations in the DNMT3A DNA methyltransferase in acute myeloid leukemia patients cause both loss and gain of function and differential regulation by protein partners
Sandoval JE, Huang YH, Muise A, Goodell MA, Reich NO. J. Bio. Chem., 294(13) (2019), 4898-4910
https://doi.org/10.1074/jbc.RA118.006795
Shape Matters: Gold Nanoparticle Shape Impacts the Biological Activity of siRNA Delivery
Morgan E, Wupperfeld D, Morales DP, Reich NO. Bioconjugate Chem., 30(3) (2019), 853-860
https://doi.org/10.1021/acs.bioconjchem.9b00004
2018 Publications
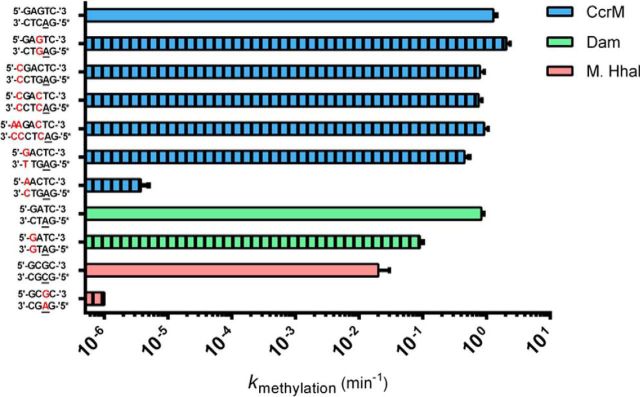
The highly specific, cell cycle\[Dash]regulated methyltransferase from Caulobacter crescentus relies on a novel DNA recognition mechanism
Reich NO, Dang E, Kurnik M, Pathuri S, Woodcock CB. J. Biol. Chem., 293(49) (2018), 19038-19046
https://doi.org/10.1074/jbc.RA118.005212
2017 Publications
Caulobacter crescentus Cell Cycle-Regulated DNA Methyltransferase Uses a Novel Mechanism for Substrate Recognition
Woodcock CB, Yakubov AB, Reich NO. Biochemistry-US, 56(30) (2017), 3913-3922
https://doi.org/10.1021/acs.biochem.7b00378
Affinity-Based Assembly of Peptides on Plasmonic Nanoparticles Delivered Intracellularly with Light Activated Control
Morales DP, Wonderly WR, Huang X, McAdams M, Chron AB, Reich NO. Bioconjugate Chem., 28(7) (2017), 1816-1820
https://doi.org/10.1021/acs.bioconjchem.7b00276
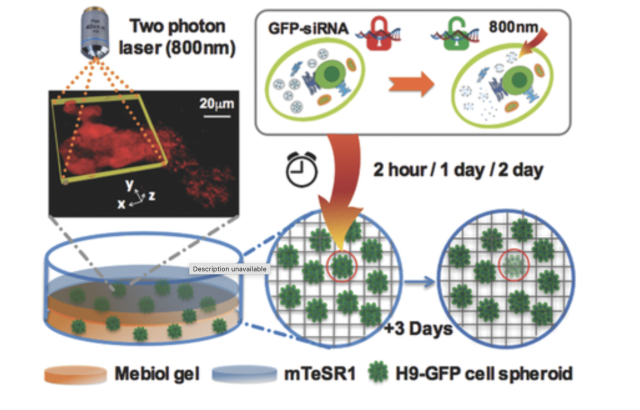
2016 Publications
Light-Patterned RNA Interference of 3D-Cultured Human Embryonic Stem Cells
Huang X, Hu Q, Lai Y, Morales DP, Clegg DO, Reich NO. Adv. Mater., 28(48) (2016), 10732-10737
https://doi.org/10.1002/adma.201603318
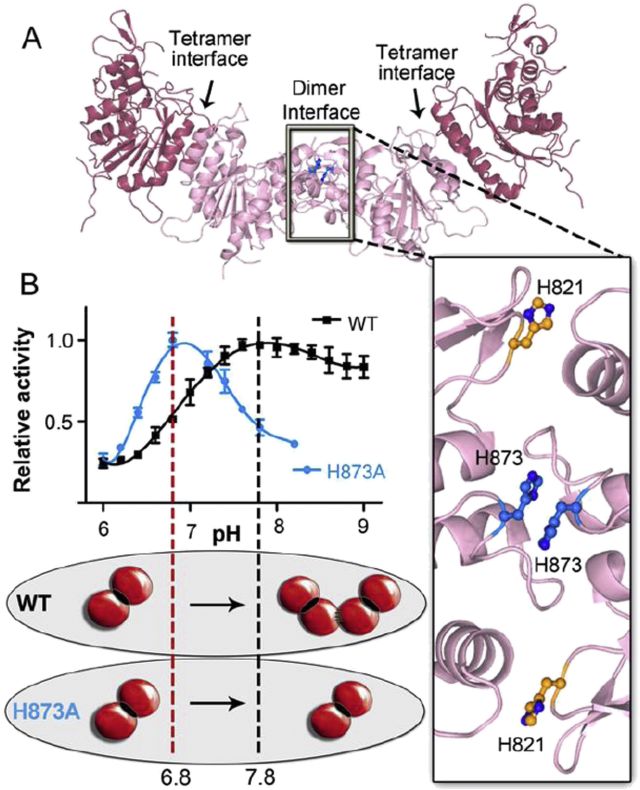
2015 Publications
De novo DNA methyltransferase DNMT3A: Regulation of oligomeric state and mechanism of action in response to pH changes
Holz-Schietinger C, Reich NO. BBA-Gen. Subjects, 1850(6) (2015), 1131-1139
https://doi.org/10.1016/j.bbagen.2015.02.003
Targeted Intracellular Delivery of Proteins with Spatial and Temporal Control
Morales DP, Braun GB, Pallaoro A, Chen R, Huang X, Zasadzinski JA, Reich NO. Mol. Pharm., 12(2) (2015), 600-609
https://doi.org/10.1021/mp500675p
Light-activated RNA interference in human embryonic stem cells
Huang X, Hu Q, Braun GB, Pallaoro A, Morales DP, Zasadzinski JA, Clegg DO, Reich NO. Biomaterials, 63 (2015), 70-79
https://doi.org/10.1016/j.biomaterials.2015.06.006
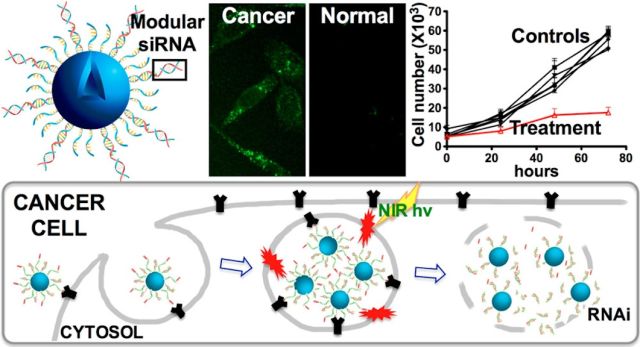
2014 Publications
Modular Plasmonic Nanocarriers for Efficient and Targeted Delivery of Cancer-Therapeutic siRNA
Huang X, Pallaoro A, Braun GB, Morales DP, Ogunyankin MO, Zasadzinski JA, Reich NO. Nano Lett., 14(4) (2014), 2046-2051
https://doi.org/10.1021/nl500214e
2012 Publications
RNA modulation of the human DNA methyltransferase 3A
Holz-Schietinger C, Reich NO. Nucleic Acids Res., 40(17) (2012), 8550-8557
https://doi.org/10.1093/nar/gks537
Mutations in DNA Methyltransferase (DNMT3A) Observed in Acute Myeloid Leukemia Patients Disrupt Processive Methylation
Holz-Schietinger C, Matje DM, Reich NO. J. Bio. Chem., 287(37) (2012), 30941-30951
https://doi.org/10.1074/jbc.M112.366625
2011 Publications
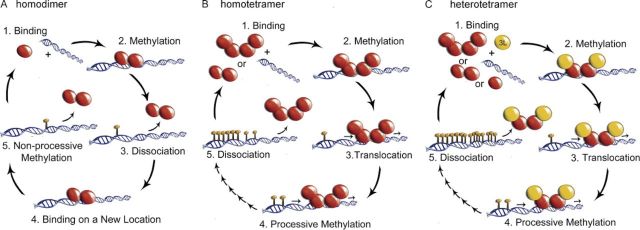
Oligomerization of DNMT3A Controls the Mechanism of de Novo DNA Methylation
Holz-Schietinger C, Matje DM, Harrison MF, Reich NO. J. Bio. Chem., 286(48) (2011), 41479-41488
https://doi.org/10.1074/jbc.M111.284687
2010 Publications
Identification of a second DNA binding site in human DNA methyltransferase 3A by substrate inhibition and domain deletion
Purdy MM, Holz-Schietinger C, Reich NO. Arch. Biochem. Biophys., 498(1) (2010), 13-22
https://doi.org/10.1016/j.abb.2010.03.007
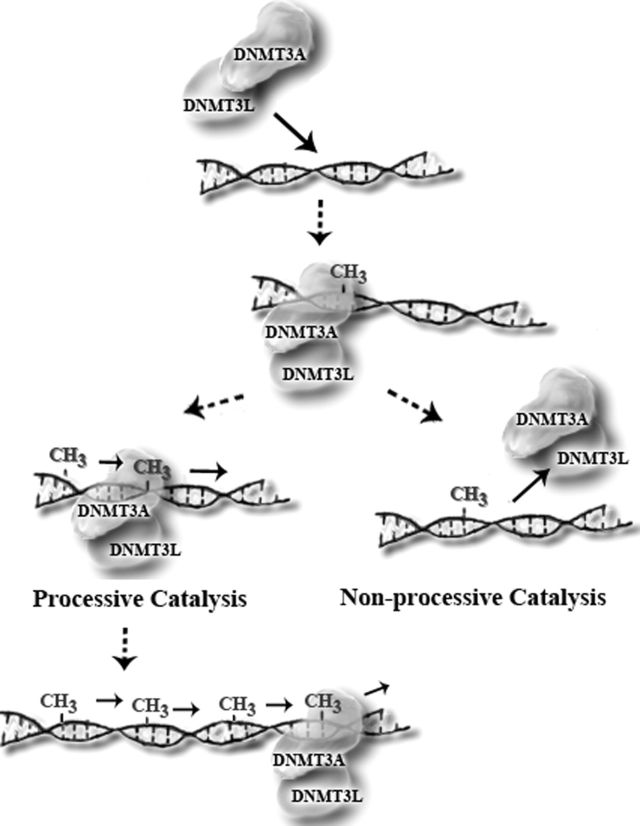
The Inherent Processivity of the Human de Novo Methyltransferase 3A (DNMT3A) Is Enhanced by DNMT3L
Holz-Schietinger C, Reich NO. J. Biol. Chem., 285(38) (2010), 29091-29100
https://doi.org/10.1074/jbc.M110.142513

2008 Publications
Enzyme-Directed Positioning of Nanoparticles on Large DNA Templates
Braun GB, Dietchtierow M, Wilkinson S, Schmidt F, Hüben M, Weinhold E, Reich NO. Bioconjugate Chem., 19(2) (2008), 476-479
https://doi.org/10.1021/bc700275h

2007 Publications
Specific and sensitive detection of nucleic acids and RNases using gold nanoparticle\[Dash]RNA\[Dash]fluorescent dye conjugates
Kim JH, Estabrook RA, Braun GB, Lee BR, Reich NO. Chem. Commun., (42) (2007), 4342
https://doi.org/10.1039/b710306a
Chemically Patterned Microspheres for Controlled Nanoparticle Assembly in the Construction of SERS Hot Spots
Braun GB, Pavel I, Morrill AR, Seferos DS, Bazan GC, Reich NO, Moskovits M. J. Am. Chem. Soc., 129(25) (2007), 7760-7761
https://doi.org/10.1021/ja072533e
Detection of Sequence-Specific Protein-DNA Interactions via Surface Enhanced Resonance Raman Scattering
Bonham AJ, Braun GB, Pavel I, Moskovits M, Reich NO. J. Am. Chem. Soc., 129(47) (2007), 14572-14573
https://doi.org/10.1021/ja0767837
2006 Publications
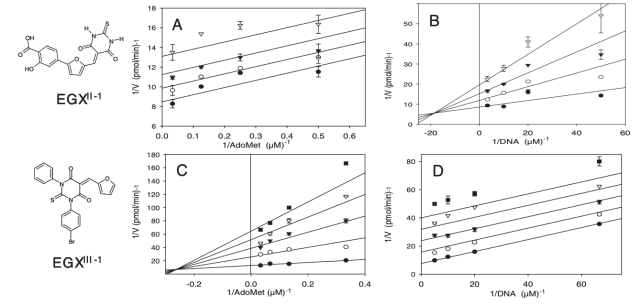
Selective Inhibitors of Bacterial DNA Adenine Methyltransferases
Mashhoon N, Pruss C, Carroll M, Johnson PH, Reich NO. J. Biomol. Screen, 11(5) (2006), 497-510
https://doi.org/10.1177/1087057106287933
2005 Publications
Controlled Spacing of Cationic Gold Nanoparticles by Nanocrown RNA
Koyfman AY, Braun GB, Magonov S, Chworos A, Reich NO, Jaeger L. J. Am. Chem. Soc., 127(34) (2005), 11886-11887
https://doi.org/10.1021/ja051144m
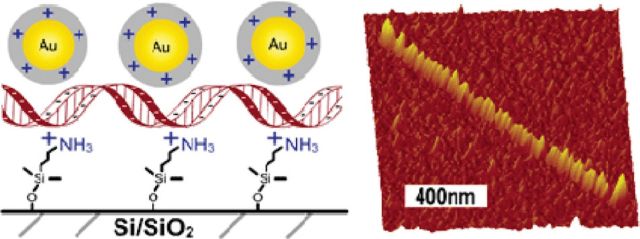
Gold Nanoparticle Decoration of DNA on Silicon
Braun GB, Inagaki K, Estabrook RA, Wood DK, Levy E, Cleland AN, Strouse GF, Reich NO. Langmuir, 21(23) (2005), 10699-10701
https://doi.org/10.1021/la0515367